医疗器械行业的研发管理,比其他制造业多了一道严苛的门槛——合规。FDA 21 CFR Part 820、ISO 13485、NMPA注册审查……每一次审查都会追问:设计变更有没有完整记录?BOM数据从哪里来、谁审批的、何时生效?在没有PDM/PLM系统支撑的企业里,这些问题往往要靠工程师翻几年前的文件夹来回答,风险极高。本文从医疗器械行业的合规需求出发,讲清楚一个能真正支撑注册审查的研发数据管理系统该具备什么能力。
医疗器械研发管理的三大特殊性
1. 设计历史文档(DHF)的完整性要求
FDA QSR和ISO 13485都要求企业建立并维护设计历史文档(Design History File,DHF),记录产品设计和开发过程的完整历史,包括:设计输入/输出、设计验证与确认记录、设计评审记录、设计变更记录。
在没有系统的情况下,DHF通常散落在各工程师的本地电脑、共享网盘、邮件附件里。一旦有人员离职,相关文件可能永久消失。审查时临时整理,不仅耗时,还存在数据不完整的法律风险。
2. 设计变更的闭环管理
医疗器械的每一次设计变更都需要评估对产品安全性和有效性的影响,记录变更理由、评估结论、审批人、生效日期,并追溯到受影响的BOM和图纸版本。
这个要求与普通制造业相比严格得多:不能只改了图纸就算完,要形成变更链——从问题发现,到变更分析,到执行,到通知,到关闭,每个节点都要有记录。
3. 物料可追溯性(Traceability)
监管机构要求企业能够追溯:哪批产品用了哪个版本的哪些零件?某个供应商提供的物料用在了哪些产品上?如果发生召回,能精确定位受影响的产品范围。
没有系统的企业,物料追溯完全依赖人工查询,速度慢、容易出错,面对召回事件时风险极大。
现实困境:没有系统时的典型痛点
某三类医疗器械企业(深圳,研发50人,产品线6条)在引入系统前的真实状态:
图纸管理:工程师各自维护本地文件夹,共享盘按项目分文件夹,但命名规则每人不同,经常找不到最新版图纸。
变更管理:变更走Word表格+邮件流转,审批完成后由文控手工更新图纸,经常有漏改的。每次审查前要专门安排1-2周时间整理DHF,加班加点。
BOM管理:机械BOM在SolidWorks,电气BOM在Excel,由文控手工合并给采购。合并一次需要半天,合并错误发生率约10%。
物料追溯:遇到客户质量投诉,需要手工查阅3年前的采购记录和生产工单,平均耗时3-5天,无法在客户要求的时间内完成响应。
系统化解法:鹏焬OIDS医疗器械方案
模块一:设计数据统一管理,DHF自动归集
鹏焬OIDS将设计过程中的所有文档(3D模型、2D工程图、电路图、测试报告、评审记录)统一存储在受控库中,每个文件自动记录:创建人、创建时间、修改人、修改内容、审批状态、当前版本号。
DHF所需的设计输入/输出文档,可以按产品、项目、版本自动汇总导出,审查时无需临时整理,随时可出具完整档案。
模块二:变更管理全流程闭环
鹏焬OIDS内置符合CMII规范的变更流程:

每个变更的来源(客户需求/设计错误/物料停产/工艺优化)在系统中分类记录,可以按季度、按类型生成变更分析报表,识别高频变更原因,推动设计质量持续改进。
模块三:BOM与物料追溯
鹏焬OIDS的BOM管理支持:
○ 设计BOM(EBOM)与制造BOM(MBOM)统一管理,自动映射
○ 每个物料关联唯一编码,记录供应商信息、批次信息
○ BOM版本历史完整保存,可查询任意历史时间点的BOM状态
○ 批次级追溯:输入任意物料编码,查询该物料用在了哪些产品批次中
面对质量投诉或召回场景,系统可在5分钟内完成物料追溯分析,精确定位受影响产品范围。
模块四:权限管控与数据安全
医疗器械企业的设计数据涉及核心知识产权和患者安全,权限管控要求严格:
○ 按角色设置访问权限(工程师/审批人/供应商/外审专家)
○ 敏感图纸支持"只读查看,不可下载"模式,防止数据外泄
○ 所有操作(查看/下载/修改/审批)留有完整日志,满足审计追溯要求
○ 外部协作(供应商/外审)通过受控链接访问,不暴露内部文件结构
实施效果数据(某三类医疗器械客户)

适合引入系统的时机
很多医疗器械企业有一个误区:等到产品线多了、团队大了再上系统。但实际上,越早建立规范的数据管理体系,历史数据积累越干净,DHF越完整,后期注册审查越顺利。
以下情况说明该考虑系统了:
○ 研发团队超过10人,图纸开始出现版本混乱
○ 有产品即将申请注册,需要整理DHF
○ 变更通知出现过遗漏,导致采购或生产错误
○ 有外部审查(FDA/NMPA/ISO 13485)在近期计划中
FAQ
Q1:鹏焬OIDS符合FDA 21 CFR Part 820的要求吗?
A:系统的变更管理、版本控制、审批记录、操作日志等功能覆盖了FDA QSR对设计控制的核心要求,已有多家三类医疗器械企业通过使用鹏焬OIDS顺利完成FDA审查准备。
Q2:医疗器械产品线很多,系统能支持多产品线并行管理吗?
A:支持。系统按项目/产品线隔离管理,每条产品线有独立的DHF、BOM版本和变更记录,互不干扰。
Q3:供应商需要查看图纸,如何控制权限?
A:可以给供应商分配受控访问账号,设置只能查看指定产品/零件的图纸,不能下载或看到其他产品资料。
Q4:现有数据(Excel BOM、共享盘图纸)如何迁移?
A:提供数据迁移服务,包括历史图纸批量导入、BOM数据清洗、版本关系重建。
Q5:系统上线需要多长时间?
A:医疗器械行业标准实施周期约2-3个月,含数据迁移、流程配置、员工培训。
如果你的企业有注册审查计划,或者正在被DHF整理、变更管理、物料追溯困扰,欢迎申请鹏焬OIDS医疗器械专项演示。
相关新闻

2026-06-25
风电装备制造企业的研发数据管理:大型装配体BOM为什么这么难管?

2026-06-24
小企业PLM系统模块化选购指南:哪些模块是必选,哪些可以等等

2026-06-23
研发数据备份靠手动,一次硬盘损坏毁掉三年积累——这件事正在很多工厂里等着发生

2026-06-22
“我改过的图纸怎么变成上周的版本了?”——4个版本覆盖事故的真实复盘
2026-06-19
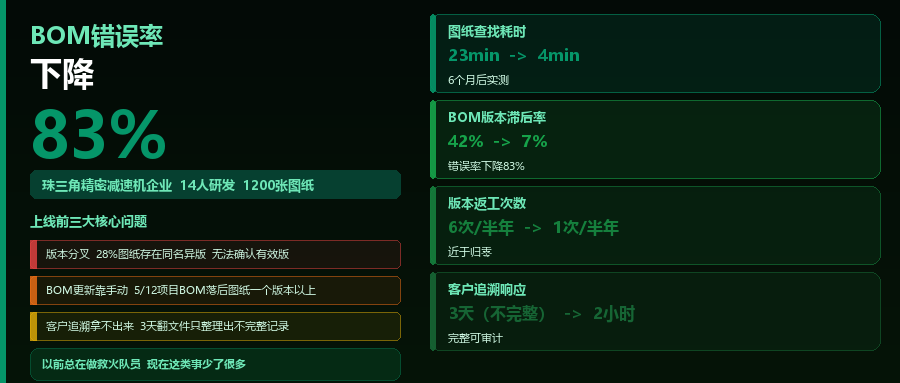
某精密机械企业:用鹏焬OIDS管住1200张图纸,BOM错误率下降83%

2026-06-18
精密机械装备制造行业PLM:多品种小批量的研发数据管理为什么这么难?

联系热线:
0755-83556155

扫一扫,关注
汉拓科技官网




Copyright © 2021 深圳市汉拓科技有限公司 粤ICP备10224947号 网站建设:万广互联






